Rothmund-Thomson Syndrome: in Search of a New Gene
Project location: Italy
Project start date: December 2007 -
Project end date: December 2008
Project number: 2007-14
Beneficiary: Università degli Studi di Milano
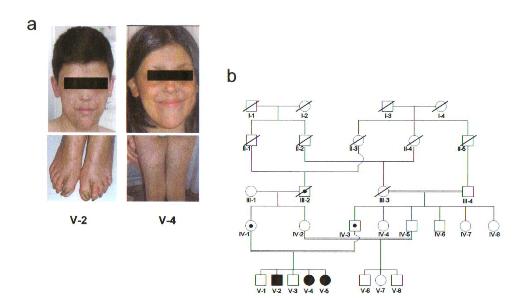
Figure 1 (a) Facial appearances of affected proband (V-2) and sib (V-4) at age 22 and 13 showing poikiloderma extending to buttocks and legs and pachyonychia of the toes. (b) Five generation pedigree of the index family: individuals affected with PN are indicated by shaded symbols and obligate carriers by dotted symbols. The inbreeding coefficient of the fifth generation sibs is 3/64
The diagnosis of Rothmund- Thomson syndrome of family members V-2, V-4 and V-5 (Fig.1a,b) was revisited as all the affected had no mutation in the RECQL4 gene and their haplotypes at the RECQL4 locus were not the same. According to a persistent neutropenia, Poikiloderma with Neutropenia (PN, OMIM %604173) has been diagnosed, a distinct subtype of hereditary poikilodermas, a clinically and genetically heterogenous group of disorders including Rothmund-Thomson syndrome (RTS, OMIM #268400).
Clericuzio type of Poikiloderma with Neutropenia, is a distinct subtype of hereditary poikilodermas, a clinically and genetically heterogenous group of disorders including Rothmund-Thomson syndrome (RTS, OMIM #268400). The disorder is characterised by specific skin manifestations, mainly a papular erythematous rash on the limbs during the first year of life which spreads centripetally and evolves into poikiloderma with hypo and hyperpigmented areas, and pachyonychia, especially of the toe nails. The most important extracutaneous sign is chronic neutropenia: a neutrophil dysfunction caused by a defect in the oxidative metabolism, and ultimately resulting in an increased susceptibility to infections and risk of leukemic transformation. Clear autosomal recessive inheritance has been supported by the occurrence of affected and non affected siblings from healthy but often consanguineous parents (Wang et al, 2003; Van Hove et al , 2005; Mostefai et al, 2008). PN shows extensive phenotypic overlap with RTS, but some phenotypic differences point towards a distinct genetic control. Mutations of the helicase gene RECQL4, which are found in two thirds of RTS patients, are consistently absent in PN patients.
Genome-wide SNP scan over all living family members (n=18) was performed on all living subjects of the last three generations using a Genechip® Human Mapping 262k NspI SNP Array (Affymetrix). Data obtained in this way, were subsequently used for two-point linkage analysis using the GeneHunter software package and assuming an autosomal recessive trait with 100% penetrance. Additional inclusion-criteria included sharing of homozygous regions among the three affected sibs, the carrier status of their parents and the exclusion of the Identical By Descent (IBD) haplotype by the maternal grandmother III-1. We trimmed the pedigree to maximize statistical power and performed quality control on SNP data discarding all ambiguous genotypes and SNPs with a call rate under an arbitrary 95% threshold.
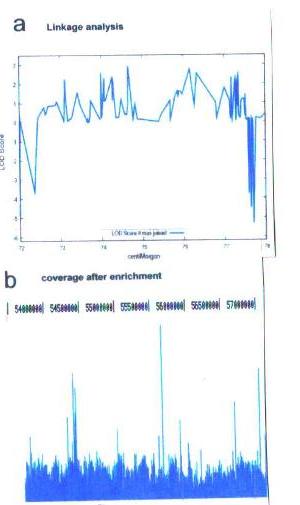
Figure 2 (a) LOD score diagram zoom-in of the ca. 5cM region (b) Post-array capture coverage of (V-2 DNA). The calculated enrichment value is 241
Linkage analysis identified three such candidate regions with a LOD score >2.5 of which we selected one region since it was the largest genomic interval where LOD score values consistently stayed above -4 (Fig.2a). The genetic size of this interval (5cM) is consistent with the pedigree, being the inbreeding coefficient directly related with the length of the region inherited IBD (Genin 1998. The locus spans 3.4 Mb, encompasses 276 consecutive SNPs consistent with linkage criteria, and contains over 80 genes. The latter fact prompted us to apply array-based sequence enrichment and next generation sequencing, a strategy enabling an unbiased search for disease associated mutations in large genomic intervals.
A genomic shotgun library from proband V-2 (Fig.1b) was prepared with paired-ends adapters following the Illumina (http://www.illumina.com) protocol, and selecting for inserts of 100 bp. Following quality control, sequence-capture was performed on a custom repeats-masked 244K array (Agilent ImaGenes GmbH, Berlin) using a 4 bp tiling between oligos on repeat-free regions by which the tagged region dropped to 1.7 Mb (Fig.2b). The final library eventually produced >7 x 106 reads of 36 bases, which were mapped onto the human reference genome using ELAND (from the Illumina GA pipeline 1.0) or Maq V0.7.1. Next generation sequencing of the target region revealed 1487 nucleotide changes in comparison to the human reference genome sequence (UCSC Build, hg18/NCBI Build 36.1 [March 2006])(Fig.1d). Subsequent analysis ultimately led to the identification of a consistent A to C substitution within a highly conserved gene.
This mutation affects a AG > CG acceptor splice site and segregates as expected across the last three generations. By direct capillary sequencing (ABI Prism 3730) we confirmed the carrier and non carrier status of the living individuals within the pedigree, and cDNA analysis revealed an aberrant transcript.
We validated the association between the gene and PN with a second case: an Italian sporadic patient diagnosed with RTS and myelodysplasia (Pianigiani et al, 2001) displaying a typical PN phenotype with no mutations at RECQL4, who was found to be a compound heterozygote for two different mutations in the same gene.
All the mutations was found to be absent in 350 Italian control chromosomes.



