Rothmund-Thomson Syndrome: in Search of a New Gene
Project location: Italy
Project start date: December 2007 -
Project end date: December 2008
Project number: 2007-14
Beneficiary: Università degli Studi di Milano
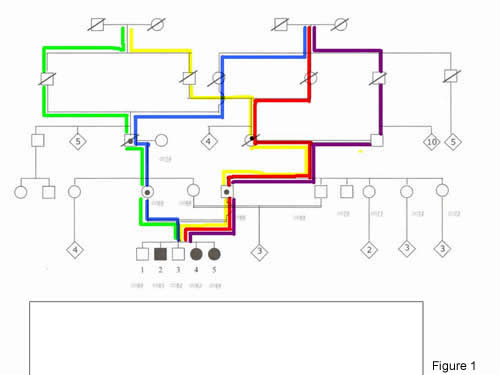 The three siblings of a five-generation inbred Italian family (Figure 1) were initially misdiagnosed with Rothmund-Thomson (RTS [MIM #268400]), a progeria-like syndrome showing phenotypic overlap with Clericuzio-type poikiloderma with neutropenia (PN [MIM %604173]). Both syndromes are rare autosomal-recessive genodermatosis characterised by poikiloderma and short stature, but differential diagnosis is based on radial defects and alopecia (RTS patients), vs pachyonychia and severe chronic neutropenia (PN patients Figure 2).
The three siblings of a five-generation inbred Italian family (Figure 1) were initially misdiagnosed with Rothmund-Thomson (RTS [MIM #268400]), a progeria-like syndrome showing phenotypic overlap with Clericuzio-type poikiloderma with neutropenia (PN [MIM %604173]). Both syndromes are rare autosomal-recessive genodermatosis characterised by poikiloderma and short stature, but differential diagnosis is based on radial defects and alopecia (RTS patients), vs pachyonychia and severe chronic neutropenia (PN patients Figure 2).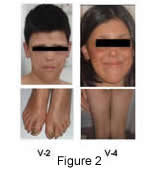
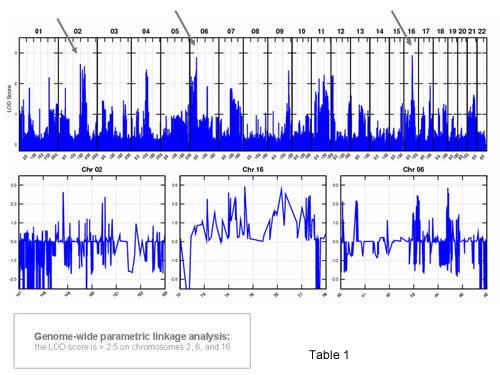 This genomic region hosts at least 80 genes, and none was a suitable candidate gene for PN. The standard process of sequencing candidate genes seemed an extremely costly and time consuming procedure. We decided to bypass this step using the new technology of targeted next generation DNA sequencing.
This genomic region hosts at least 80 genes, and none was a suitable candidate gene for PN. The standard process of sequencing candidate genes seemed an extremely costly and time consuming procedure. We decided to bypass this step using the new technology of targeted next generation DNA sequencing.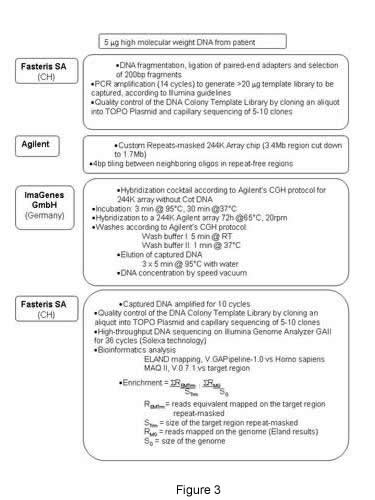 The stepwise procedure is reported (Figure 3). The output of the new sequencing method consists in two files reporting all the mismatches of the input sequence compared against public databases; one file records all the homozygous mismatches and the second the heterozygous mismatches. According to our model, we considered only the homozygous SNP file.
The stepwise procedure is reported (Figure 3). The output of the new sequencing method consists in two files reporting all the mismatches of the input sequence compared against public databases; one file records all the homozygous mismatches and the second the heterozygous mismatches. According to our model, we considered only the homozygous SNP file.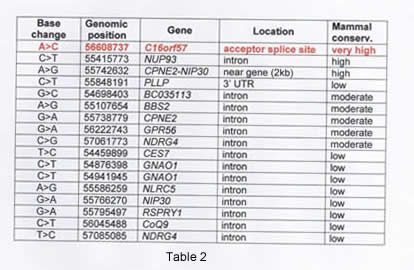
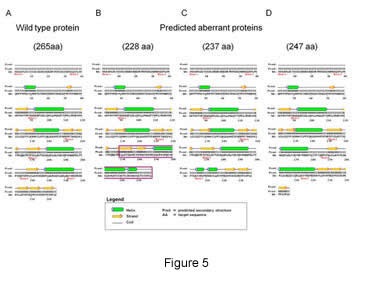
We validated the role of C16orf57 gene as a second unrelated patient with PN, who was shown to be a compound heterozygote for C16orf57 mutations (Figs. 4, 5).
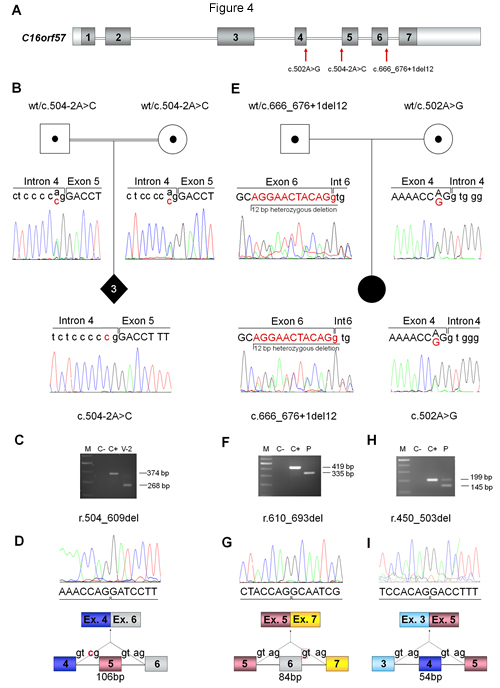
Figure 4 (above) C16orf57 Mutational Analysis
(A) Schematic genomic structure of C16orf57 gene (16q13) spanning 20Kb and 7 exons (dark boxes) encoding a 265 aa protein. Red arrows point to the identified mutations (primers and conditions are reported in Table S3).
(B and E) Pedigrees and sequencing of C16orf57 genomic mutations: (B) shows the carrier status of both parents and the homozygous splicing mutation c.504-2A>C of the three affected sibs and (E) the parental origin and the two mutations c.666_676+1del12/c.502A>G of the compound heterozygote sporadic patient. Base changes are in red characters.
(C, F, H) Agarose gel electrophoresis showing products of different RT-PCR (primers and conditions are reported in Table S4), on lymphoblastoid cell lines RNA. Lengths of normal and aberrant products are indicated. The following abbreviations are used: V-2 homozygous patient; P compound heterozygous patient; C+ normal control; C- negative control, M Gene Ruler TM DNA Ladder mix (Fermentas).
(D, G, I) Sequence data of the two PN patients showing aberrant transcripts due to out of frame skipping of exon 5, in frame skipping of exon 6 and exon 4, respectively, and related schematic diagrams of the mutation-associated miss-spliced cDNAs.
Targeted next-generation sequencing appoints c16orf57 as clericuzio-type poikiloderma with neutropenia gene. Volpi L, Roversi G, Colombo EA, Leijsten N, Concolino D, Calabria A, Mencarelli MA, Fimiani M, Macciardi F, Pfundt R, Schoenmakers EF, Larizza L.
Am J Hum. Genet. 2010 Jan;86(1):72-6. Epub 2009 Dec 10.



